GROMACS
GROMACS是一个用于分子动力学模拟的多用途软件包。它主要是为蛋白质、脂质、核酸等有很多复杂的成键交互作用的生物化学分子设计的。但是由于GROMACS在计算非成键相互作用时非常快,它也被用于聚合体等非生物系统。GROMACS支持所有现代分子动力学计算的常见算法。
用任务模式如何提交任务
一、 提交流程
登录 Fastone 平台控制台;
数据管理上传计算文件;
在首页,点击【提交作业】-点击【GROMACS】应用-选择任务模式-选择机器配置,进行启用机器;
上传计算文件,设置资源参数;
二、 单机模式
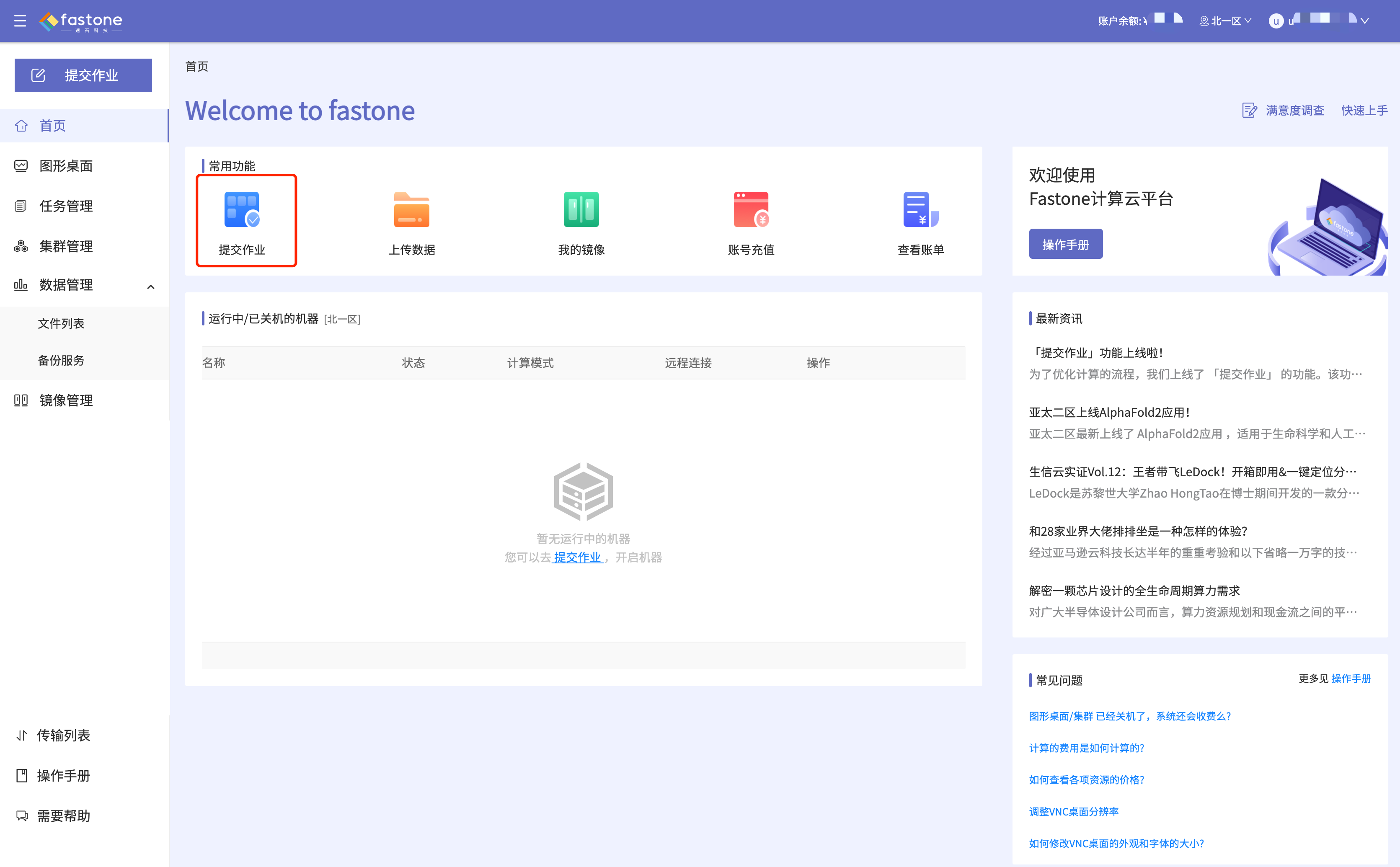
Step 1:首页选择【提交作业】,如图:
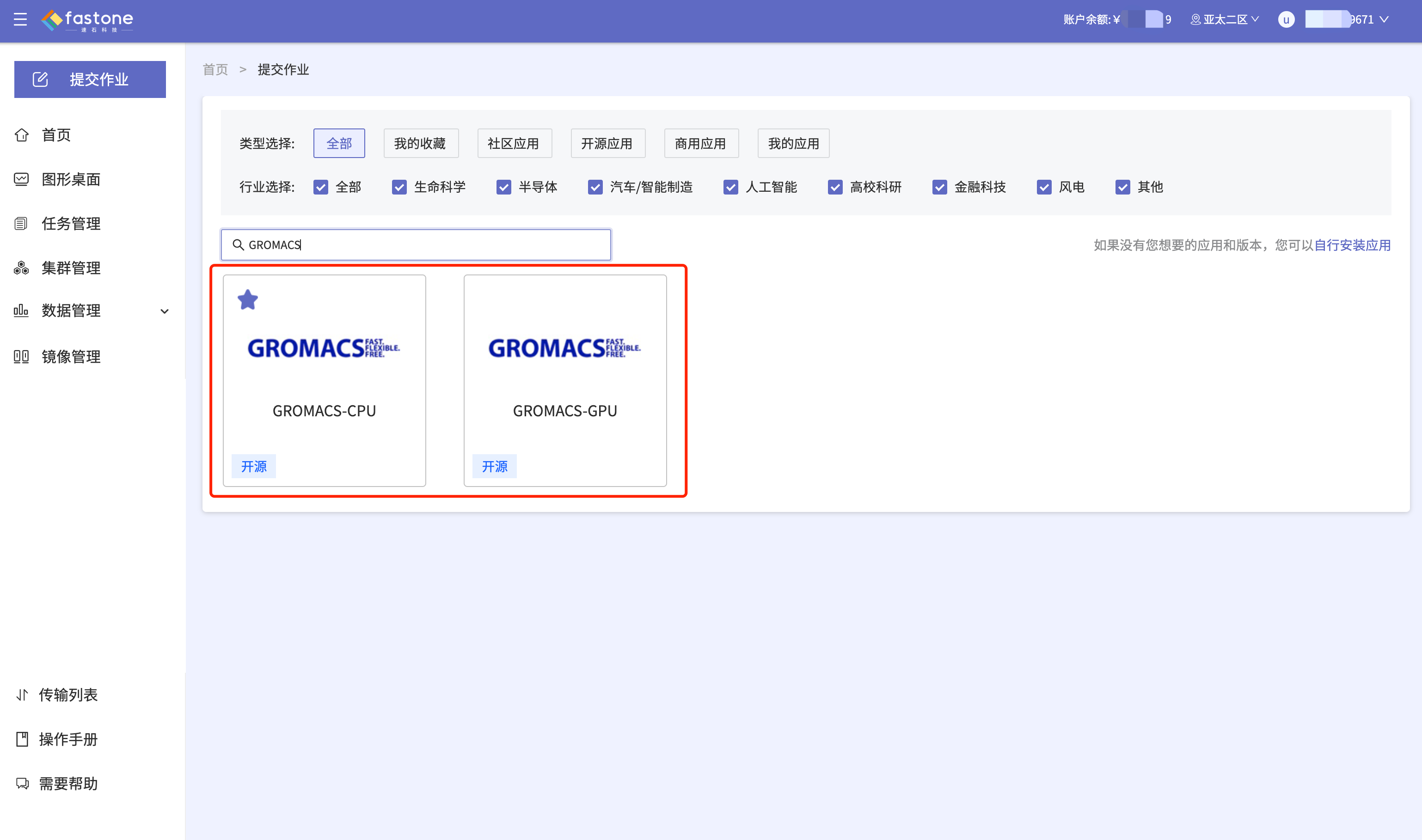
Step 2:选择应用【GROMACS】;
- 注意GROMACS-CPU和GROMACS-GPU的选择
Step 3. 上传输入文件,配置运行参数;
可以设置提交的作业名
可点击感叹号查看待上传输入文件的格式、软件运行参数的注意事项等
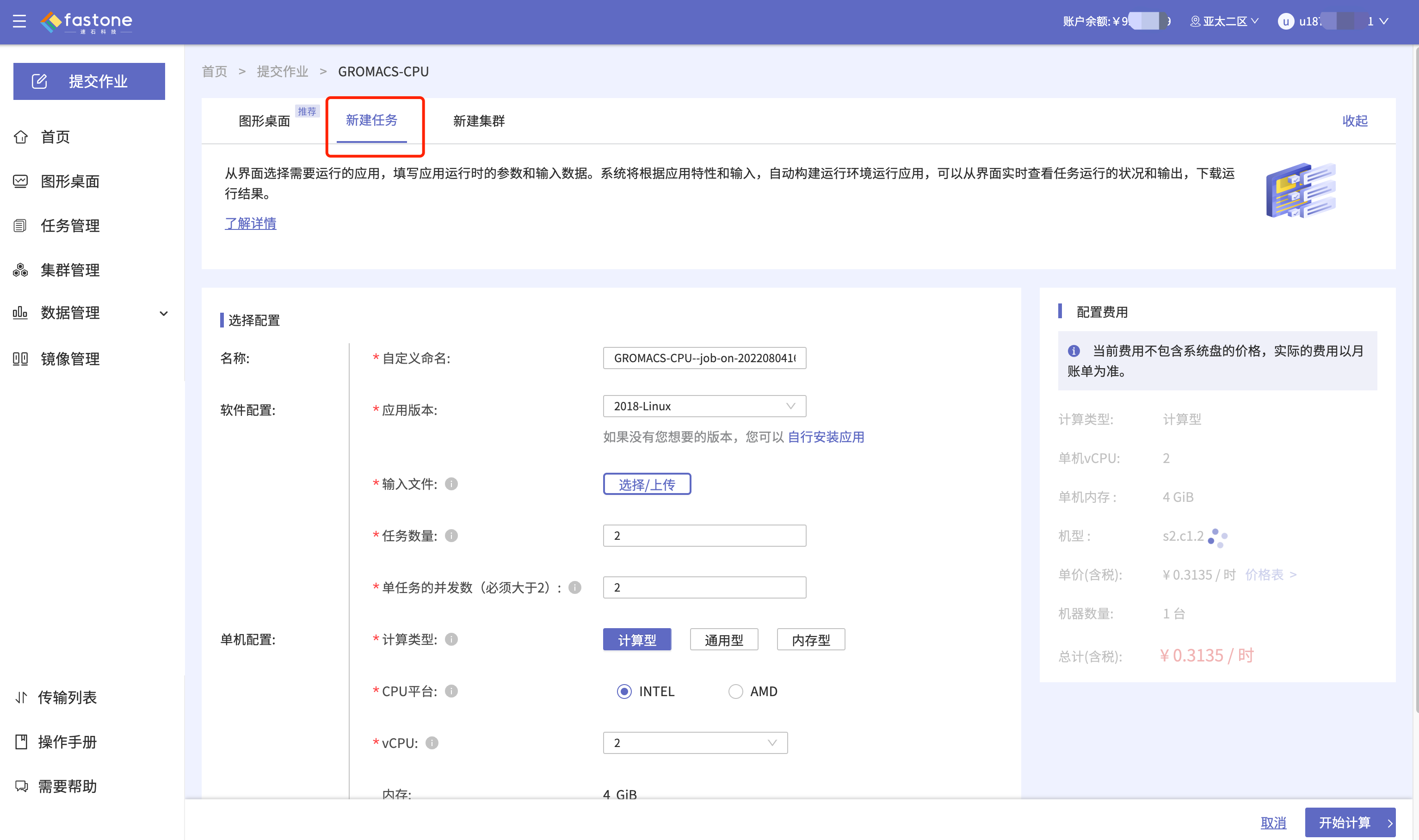
- 可以设置提交的作业名。
- 可点击感叹号查看待上传输入文件的格式、软件运行参数的注意事项等。
Step 4. 选择硬件配置;
- 根据软件版本和使用需求,选择CPU/GPU配置。
- 并发数量:单任务并发数。
- 内存:设置各个计算节点内存大小为单节点核心数×内存配比。
- 可点击感叹号查看相关设置要求。
Step 5. 查看作业内容汇总,并提交作业;
三、 提交后的监控
- 作业提交后可通过任务管理查看任务的输出和日志等内容。
- 可通过集群监控查看正在运行的计算节点资源使用率等信息。